Module 17 Complex data types in experimental data pre-processing
Raw data from many biomedical experiments, especially those that use high-throughput techniques, can be large and complex. As Vince Buffalo notes in an article on bioinformatics data skills:
“Right now, in labs across the world, machines are sequencing the genomes of the life on earth. Even with rapidly decreasing costs and huge technological advancements in genome sequencing, we’re only seeing a glimpse of the biological information contained in every cell, tissue, organism, and ecosystem. However, the smidgen of total biological information we’re gathering amounts to mountains of data biologists need to work with. At no other point in human history has our ability to understand life’s complexities been so dependent on our skills to work with and analyze data.”313
In previous modules, we have gone into a lot of detail about advantages of the tidyverse approach. However as you work with biomedical data, you may find that it is unreasonable to start with a tidyverse approach from the first steps of pre-processing the data. This is particularly the case if you are working with data from complex research equipment, like mass spectrometers and flow cytometers.
It can be frustrating to realize that you can’t use your standard tools in some steps of working with the data you collect in your experiments. For example, you may have taken an R course or workshop, and be at the point where you are starting to feel pretty comfortable with how to use R to work with standard datasets. You can feel like you’re starting at square one when you realize that approach won’t work for some steps of working with the data you’re collecting for your own research.
This module aims to help you navigate this process. In particular, it is helpful to understand how the Bioconductor approach differs from the tidyverse approach, to start developing a framework and tools for navigating both approaches.
The primary difference between the two approaches is how the data objects are structured. When you work with data in R, it is kept in an “object”, which you can think of as a structured container for the data. In the tidyverse approach, the primary data container is the dataframe. A dataframe is made up of a set of object types called vectors: each column in the dataframe is a vector. Therefore, to navigate the tidyverse approach, the only data structures you need to understand well are the dataframe structure and the vector structure. Tools in the tidyverse use these simple structures over and over.
By contrast, the Bioconductor approach uses a collection of more complex data containers. There are a number of reasons for this, which we’ll discuss in this module.
As a note, it is possible that in the near future, all steps of even complex pipelines will be manageable with a tidyverse approach. More R developers are embracing the tidyverse approach and making tools and packages within its framework. In some areas with complex data, there have been major inroads to allow a tidyverse approach throughout the pipeline even when working with complex data. We’ll end the module by discussing these prospects.
Objectives. After this module, the trainee will be able to:
- Explain why R software for pre-processing biomedical data often stores data in complex, “untidy” formats
- Describe how these complex data formats can create barriers to laboratory-based researchers seeking to use reproducibility tools for data pre-processing
- Explain how the tidyverse and Bioconductor approaches differ in the data structures they use
17.1 How the Bioconductor and tidyverse approaches differ
The heart of the difference between the tidyverse and Bioconductor approaches comes down to how data are structured within pipelines. While there are more differences than this one, most of the other differences result from this main one.
As we’ve described in detail in modules 3 and 16, in the tidyverse approach, data are stored throughout the pipeline in a dataframe structure. These dataframes are composed of a data structure called a vector. Vectors make up the columns of a dataframe. Almost every function in the tidyverse, therefore, is designed to input either a dataframe or a vector. And almost every function is designed to output the same type of data container (dataframe or vector) that it inputs. As a result, the tidyverse approach allows you to combine functions in different orders to tackle complex processes through a chain of many small steps.
By contrast, most packages in Bioconductor use more complex data structures to store data. Often, a Bioconductor pipeline will use different data structures at different points in its pipeline. For example, your data might be stored in one type of a data container when it’s read into R, and another type once you’ve done some pre-processing.
As a result, with the Bioconductor approach, there will be more types of data structures that you will have to understand and learn how to use. Another result is that the functions that you use in your pipeline will often only work with a specific data structure. You therefore will need to keep track of which type of data structure is required as the input to each function.
This also means that you are more constrained in how you chain together different functions to make a pipeline. To be clear, a pipeline in R that includes these complex Bioconductor data structures will typically still be modular, in the sense that you can adapt and separate specific parts of the pipeline. However, they tend to be much less flexible than pipelines developed with a tidyverse approach. The data structure changes often, with certain functions outputing a data structure that is needed for the next step, then the function of the next step outputting the data in a different structure, and so on. This changing data structure means that the functions for each step often are constrained to always be put in the same order. By comparison, the small tools that make up tidyverse functions can often be combined in many different orders, letting you build a much larger variety of pipelines with them. Also, many of the functions that work with complex data types will do many things within one function, so they can be harder to learn and understand, and they are often much more customized to a specific action, which means that you have to learn more functions (since each does one specific thing).
This difference will also make a difference in how you work when you modify a pipeline of code. In the tidyverse approach, you will change the functions you include and the order in which you call them, rearranging the small tools to create different pipelines. For a Bioconductor pipeline, it’s more common that to customize it, you will adjust parameter settings within functions, but will still call a standard series of functions in a standardized order.
Because of those differences, it can be hard to pick up the Bioconductor approach if you’re used to the tidyverse approach. However, Bioconductor is critical to learn if you are working with many types of biomedical data, as many of the key tools and algorithms for genomic data are shared through that project. This means that, for many biomedical researchers who are now generating complex, high-throughput data, it is worth learning how to use complex data structures in R.
17.2 Why is the Bioconductor approach designed as it is?
To begin learning the Bioconductor approach, it can be helpful to understand why it’s designed the way it is. First, there are some characteristics of complex data that can make the data unsuitable for a tidyverse approach, including data size and complexity. In the next section of this module, we’ll discuss some of these characteristics, as well as provide examples of how biomedical data can have them. However, there are also some historical and cultural reasons for the Bioconductor design. It is helpful to have an introduction to this, as it can help you navigate as you work within the Bioconductor framework.
Bioconductor predates the tidyverse approach. In fact, it has been around almost as long as R itself—the first version of R was first released in 2000, and Bioconductor started in 2003. The Bioconductor project was inspired by an ambitious aim—to allow people around the world to coordinate to make tools for pre-processing and analyzing genomic and other high-throughput data. Anyone is allowed to make their own extension to R as a package, including a Bioconductor package.
Imagine how complex it is to try to harness all these contributions. Within the Bioconductor project, this challenge is managed by using some general design principles, centered on standard data structures. The different Bioconductor data structures, then, were implemented to help many people coordinate to make software extensions to R to handle complex biomedical data. As Susan Holmes and Wolfgang Huber note in their book Modern Statistics for Modern Biology, “specialized data containers … help to keep your data consistent, safe and easy to use.”314 Indeed, in an article on software for computational biology, Robert Gentleman—one of the developers of R and founders of the Bioconductor project—is quoted as saying:
“We defined a handful of data structures that we expected people to use. For instance, if everybody puts their gene expression data into the same kind of box, it doesn’t matter how the data came about, but that box is the same and can be used by analytic tools. Really, I think it’s data structures that drive interoperability.” — Robert Gentlemen as quoted in Stephen Altschul et al.315
Each person who writes code for Bioconductor can use these data structures, writing functions that input and output data within these defined structures. If they are working on something where there isn’t yet a defined structure, they can define new ones within their package, which others can then use in their own packages.
As a result of this design, there are a number of complex data structures in use within Bioconductor packages. Some that you might come across as you start to work using this approach include:316
ExpressionSetSummarizedExperimentGRangesVCFVRangesBSgenome
17.3 Why is it sometimes necessary to use a Bioconductor approach with biomedical data
For data collected from complex laboratory equipment like flow cytometers and mass spectrometers, there are two main features that make it useful to use more complex data structures in R in the earlier stages of pre-processing the data rather directly using a tidy data structure. First, the data are often very large, in some cases so large that it is difficult to read them into R. Second, the data might combine various elements that you’d like to keep together as you move through the steps of pre-processing the data, but each of these elements have their own natural structures, making it hard to set the data up as a two-dimensional dataframe. Let’s take a more detailed look at each of these.
First, very large datasets are common in biomedical data, including genomics data. In a book on Modern Statistics for Modern Biology, Holmes and Huber describe how the size of biological data have exploded:
“Biology, formerly a science with sparse, often only qualitative data, has turned into a field whose production of quantitative data is on par with high energy physics or astronomy and whose data are wildly more heterogeneous and complex.”317
When datasets are large, it can cause complications for computers. A computer has several ways that it can store data. The primary storage is closely connected with the computer’s processing unit, where calculations are made, and so data stored in this primary storage can be processed by code very quickly. This storage is called the computer’s random access memory, or RAM. R uses this approach, and so when you load data in R to be stored in one of its traditional data structures, that data is moved into part of the computer’s RAM.318
Data can also be stored in other devices on a computer, including hard drives and solid state drives that are built into the computer or even onto storage devices that can be removed from the computer, like USB drives or external hard drives. The size of available storage in these devices tends to be much, much larger than the storage size of the computer’s RAM. However, it takes longer to access data in these secondary storage devices because they aren’t directly connected to the processor, and instead require the data to move into RAM before it can be accessed by the processor, which is the only part of the computer that can do things to analyze, modify, or otherwise process the data.
The traditional dataframe structure in R is built after reading data into RAM. However, many biological experiments now create data that is much too large to read into memory for R in a reasonable way.319 If you try to read in a dataset that’s too large for the RAM, R can’t handle it. As Roger Peng notes in R Programming for Data Science:
“Reading in a large dataset for which you do not have enough RAM is one easy way to freeze up your computer (or at least your R session). This is usually an unpleasant experience that usually requires you to kill the R process, in the best case scenario, or reboot your computer, in the worst case.”320
More complex data structures can allow more sophisticated ways to handle massive data, and so they are often necessary when working with massive biological datasets, particularly early in pre-processing, before the data can be summarized in an efficient way. For example, a more complex data structure could allow much of the data to be left on disk, and only read into memory on demand, as specific portions of the data are needed.321 This approach can be used to iterate across subsets of the data, only reading parts of the data into memory at a time.322 Such structures can be designed to work in a way that, if you are the user, you won’t notice the difference in where the data is kept (on disk versus in memory)—this means you won’t have to worry about these memory management issues, but instead can just gain from everything going smoothly, even as datasets get very large.323
The second reason that tidy dataframes aren’t always the best container for biomedical data has to do with the complexity of the data. Dataframes are very clearly and simply organized. However, they can be too restrictive in some cases. Sometimes, you might have data that do not fit well within the two-dimensional, non-ragged structure that is characteristic of the dataframe structure.
For example, some biomedical data may have data that records characteristics at several levels of the data. It may have records on the levels of gene expression within each sample, separate information about each gene that was measured, and another set of information that characterizes each of the samples. While it is critical to keep “like” measurements aligned with data like this—in other words, to insure that you can connect the data that characterizes a gene with the data that provides measures of the level of expression of that gene in each sample—these data do not naturally have a two-dimensional structure and so do not fit naturally into a dataframe structure.
Finally, one of the advantages of these complex data structures for biomedical data pre-processing is that they can be leveraged to develop very powerful algorithms for working with complex biomedical data. These include reading data in from the specialized file formats that are often output by laboratory equipment.324
17.5 Combining Bioconductor and tidyverse approaches in a workflow
Work with research data will typically require a series of steps for pre-processing, analysis, exploration, and visualization. Collectively, these form a workflow or pipeline for the data analysis. With large, complex biological data, early steps in this workflow might require a Bioconductor approach, given the size and complexity of the data, or because you’d like to use a method or algorithm available through Bioconductor. However, this doesn’t mean that you must completely give up the power and efficiency of the tidyverse approach described in earlier modules.
Instead, you can combine the two approaches in a workflow like that shown in Figure 17.1. In this combined approach, you start the workflow in the Bioconductor approach and transition when possible to a tidyverse approach, transitioning by “tidying” from a more complex data structure to a simpler dataframe data structure along the way. This is a useful approach, because once your workflow has advanced to a stage where it is straightforward to store the data in a a dataframe, there are a large advantages to shifting into the tidyverse approach as compared to using more complex object-oriented classes for storing the data, in particular when it comes to data analysis and visualization at later stages in your workflow. In this section, we will describe how you can make this transition to create a combined workflow.
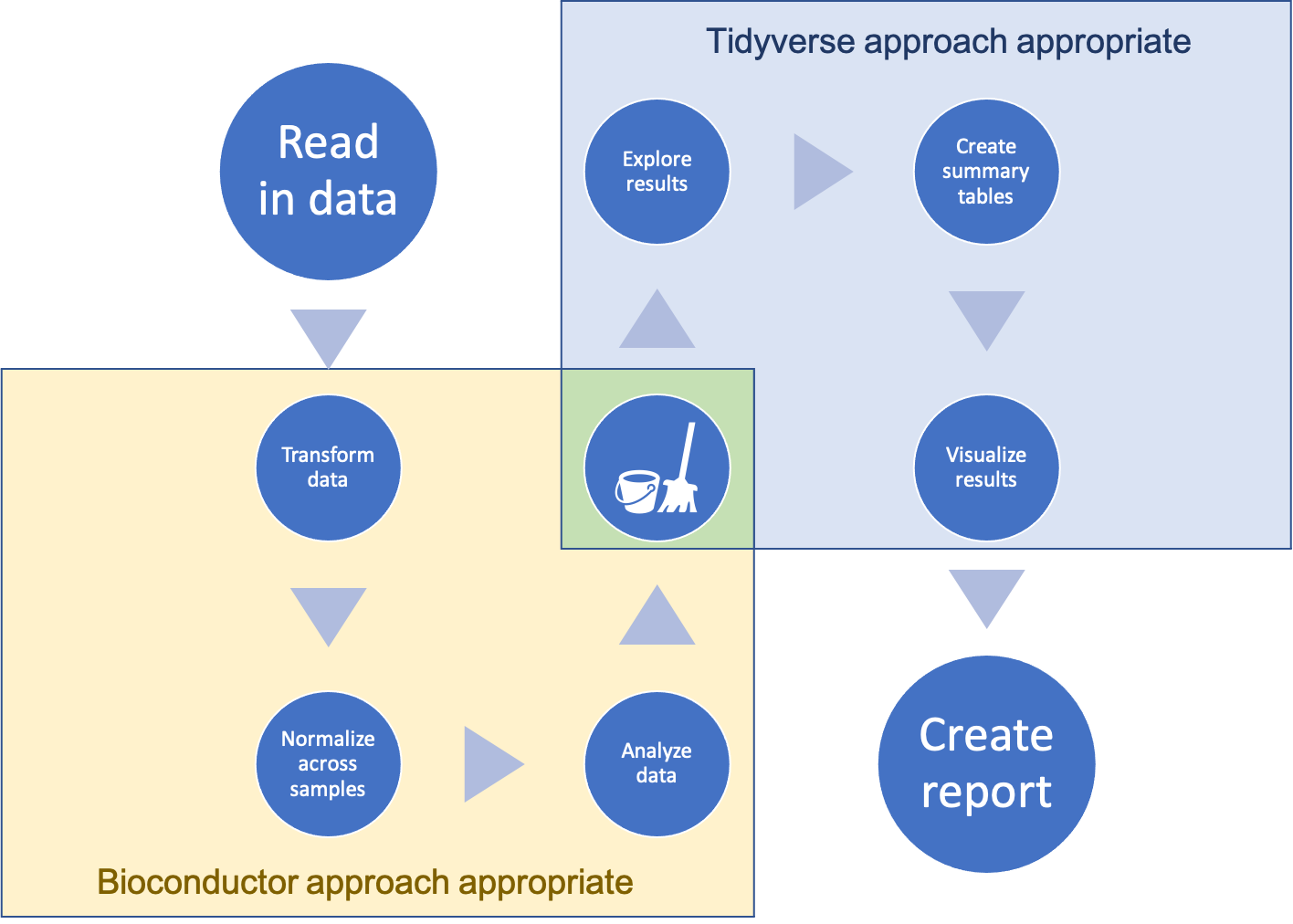
Figure 17.1: An overview of a workflow that moves from a Bioconductor approach—for pre-processing of the data—through to a tidyverse approach one pre-processing has created smaller, simpler data that can be reasonably stored in a dataframe structure.
Key to a combined pipeline are tools that can convert between specialized data
structures for Bioconductor and tidy dataframes. A set of tools for doing this
are available through the biobroom package.325 The biobroom package
includes three generic functions (also called “methods”), which can be used
on a number of Bioconductor object classes. When applied to object stored in one
of these Bioconductor classes, these functions will extract part of the data
into a tidy dataframe format. In this format, it is easy to use the tools from
the tidyverse to further explore, analyze, and visualize the data.
The three generic functions of biobroom are called tidy, augment,
and glance. These function names mimic the names of the three main functions
in the broom package, which is a more general purpose package for extracting
tidy datasets from more complex R object containers.326 The
broom package focuses on the output from functions in R for statistical
testing and modeling, while the newer biobroom package replicates this idea,
but for many of the common object classes used to store data through
Bioconductor packages and workflows.327
As an example, we can talk about how the biobroom package can be used to
convert output generated by functions in the edgeR package. The edgeR
package is a popular Bioconductor package that can be used on gene expression
data, to explore which genes are expressed differently across experimental
groups (an approach called differential expression analysis).328 Before
using the functions in the package, the data must be pre-processed to align
sequence reads from the raw data and then to create a table with the counts of
each read at each gene across each sample. The edgeR package includes
functions for pre-processing these data, including filtering out genes with low
read counts across all samples and applying model-based normalization across
samples to help handle technical bias, including differences in sequencing depth.329
The edgeR package operates on data stored in a special object class defined by
the package called the DGEList object class.330 This object
class includes areas for storing the table of read counts, in the form of a
matrix appropriate for analysis by other functions in the package, as well as
other spots for storing information about each sample and, if needed, a space to
store annotations of the genes.331 Then functions from the edgeR
package can perform differential expression analysis on the data in the
DGEList class. The result is an object in the DGEExact class, which is also
defined by the edgeR package. To extract data from this class in a tidy
format, you can use the tidy and glance functions from biobroom.
17.6 Outlook for a tidyverse approach to biomedical data
Finally, tools are continuing to evolve, and it’s quite possible that in the future there might be tidy dataframe formats that are adaptable enough to handle earlier stages in the data pre-processing for genomics data. The tidyverse dataframe approach has already been adapted to enable tidy dataframes to include more complex types of data within certain columns of the data frame as a special list-type column.
This functionality is being leveraged through the sf
package, for example, to enable a tidy approach to working with geographical
data. This allows those who are working with geographical data, for example data
from shapefiles for creating maps, to use the standard tidyverse approaches
while still containing complex data needed for this geographical information
within a tidy dataframe. Another example is the tidymodels package, which …
It seems very possible that similar approaches may be adapted in the near future to allow for biomedical or genomic data to be stored in a way that both accounts for complexity early and pre-processing of these data but also allows for a more natural integration with the wealth of powerful tools available through the tidyverse approach.